
Posters will be available after the conference in November. Email digitalcommons@providence.org if you would like to be notified when the collection is updated.
-

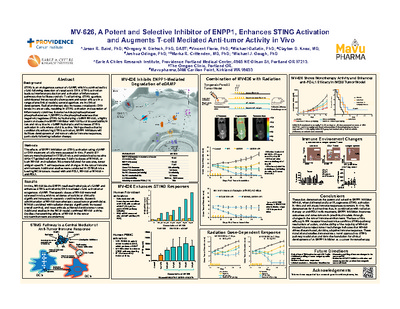
MV-626, a potent and selective inhibitor of ENPP1 enhances STING activation and augments T-cell mediated anti-tumor activity in vivo
Jason Baird, Gregory Dietsch, Vincent Florio, Michael Gallatin, Clayton Knox, Joshua Odingo, Marka Crittenden, and Michael J. Gough
Background: STING is an endogenous sensor of cGAMP, which is synthesized by cGAS following detection of cytoplasmic DNA. STING activation leads to interferon production and activation of inflammatory pathways that facilitate cytolytic T cell priming. STING agonists administered intratumorally show potent anti-tumor efficacy in a range of preclinical models; several agonists are in clinical development. Radiation therapy also increases cytoplasmic DNA levels in cancer cells, resulting in STING activation and secretion of inflammatory cytokines. Ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) is the phosphodiesterase that negatively regulates STING by hydrolyzing cGAMP. MV-626, a highly potent and selective ENPP1 inhibitor with 100% oral bioavailability in rats and mice, blocks cGAMP hydrolysis and increases STING activation in cells where cGAS is active. We hypothesize that by conditionally enhancing STING activation, ENPP1 inhibitors will facilitate development of anti-tumor cellular immune responses, particularly following radiation therapy.
Methods: The effects of ENPP1 inhibition on STING activation using cGAMP or DNA treatment of cells were assessed. Panc02-SIY tumors were implanted in C57BL/6 mice and randomized to receive 20Gy CT-guided radiation therapy, 5 daily ip doses of MV-626, or both MV-626 and radiation. Mice were followed for outcome, tumor antigen specific T cell responses and changes in the tumor immune environment. Additional studies were conducted in mice bearing MC38 tumors.
Results: In vitro, MV-626 blocks ENPP1-mediated hydrolysis of cGAMP and enhances STING activation by DNA-mediated cGAS activation or exogenous cGAMP. Therapeutic doses of MV-626 were well tolerated in mice, with no evidence of toxicity or clinically-significant increases in systemic cytokine levels. Systemic administration of MV- 626 monotherapy caused tumor growth delay. MV-626 combined with radiation therapy significantly increased overall survival, and most animals achieved durable tumor cures. Additional studies in the MC38 model confirmed MV-626 activity. Studies characterizing effects of MV-626 in the tumor microenvironment are underway.
Conclusions: These data demonstrate that a potent, selective ENPP1 inhibitor augments STING activation and enhances immune responses to tumors. We demonstrate for the first time that, in combination with radiation therapy, ENPP1 inhibition improves outcomes and cures tumors in preclinical models through changes in the tumor immune environment. These translational studies represent a novel approach to enhancing tumor directed immune response following radiation, and provide a foundation for clinical development of an ENPP1 inhibitor as a cancer immunotherapy.
-

Integrative spatially-resolved, high-plex digital profiling enables characterization of complex immune biology in the tumor microenvironment of mesothelioma
Carmen Ballesteros-Merino, Moritz Widmaier, Sarah Church, Thomas Herz, Alexei Budco, Das Medrikova, Ivan Kanchev, Andrew White, Douglas Hinerfeld, Shawn Jensen, John Handy, Rachel Sanborn, Carlo Bifulco, Sarah Warren, Joseph Beechem, and Bernard .. Fox
Background: Malignant mesothelioma is an aggressive cancer with poor prognosis and few effective therapies. Since mesothelioma is derived from the mesothelium of the lung, we hypothesize that immune cells in the tumor microenvironment (TME) may behave differently than other solid tumors. In our previous studies, utilizing multi-plexed immunofluorescence, we did not find immune phenotypes associated with improved patient survival. Here we describe a novel combination of two technologies to spatially characterize the interface between mesothelioma cells, stroma and immune cells in the TME in a high-plex capacity.
Methods: Ten FFPE mesothelioma tumors were characterized by Definiens’ Immune-Oncology Profiling (IOP) and NanoString Digital Spatial Profiling (DSP). Three alternating sequential sections were stained with Definiens’ IOP (CD8/PD-1/FOXP3, CD68/PD-L1/CD3, Granzyme B). Definiens analysis was combined to identify localization of each marker in the tumor center, invasive margin or stroma. Twelve regions-of-interest (ROIs) were then selected based on the Definiens analysis for high-plex analysis on DSP on the interleaving slide: 4 CD68-enriched, 6 CD8- enriched and 2 CD3-low. For DSP analysis, each slide was stained with a combination of fluorescent-labeled antibodies (pan-cytokeratin, CD3, CD68) and a panel of 38-antibodies each conjugated to a unique UV-photocleavable DNA barcode. ROIs from Definiens’ defined analysis were overlayed on DSP fluorescent scans, followed by UV excitation of the defined ROIs, which releases the DNA barcodes for downstream quantitation on the NanoString nCounter® platform.
Results: We found strong correlation between Definiens and NanoString analysis of T cell and macrophage markers in selected regions. Generally, patients with longer survival (>6 months) had increased density of immune infiltrates including higher density of T cells, T-cell activation markers (PD-1), higher cytokeratin levels and decreased Ki67 in the tumor center and increased tertiary lymphoid structure makers (B cells) in the invasive margin. Furthermore, STING and VISTA were highly expressed across all mesotheliomas. However, the patient with the longest survival (>31 months) expressed an immune-excluded phenotype. Co-localization analysis revealed that high CD68 density was tightly correlated to PD-L1 expression and in at least one case additional suppressive macrophage markers, including CD163 and B7-H3.
Conclusions: Already this small data set demonstrates that integration of two novel high-plex spatial analysis techniques separates distinct immune mechanisms in the TME. Our analysis suggests that macrophages are highly associated with expression of immune-inhibitory signals in mesothelioma. Therefore, we hypothesize that analysis of additional mesotheliomas may guide the development of combination immunotherapy trials that will be effective against this incurable disease.
-

Delayed immune-related events after discontinuation of immunotherapy – DIRE syndrome?
Marcus Couey, R. Bryan Bell, Ashish Patel, Marka R Crittenden, Brendan Curti, and Rom Leidner
Background: Although the temporality of immune-related adverse events (irAE) is well-recognized during immunotherapy to be highly variable and often delayed,[1] post-immunotherapy irAE are rarely described and potentially under-recognized. In 2013, two cases were reported in abstract form in Deutschen Dermatologischen Gesellschaft.[2] In July 2018 a case of autoimmune hepatitis eight months post-immunotherapy was reported in The Oncologist[3] and a dermatologic series appeared online in JAMA Dermatology.[4] With expanding indications for IO and an increasing number of clinical trials in the curative-neoadjuvant setting, larger numbers of patients are being treated in earlier stages of disease and often for short courses. Given this trend, under-recognition of delayed immune-related events (DIRE) after completion of immunotherapy could pose a growing clinical hazard.
Methods: We performed a literature review in PubMed and Google Scholar (search terms included in Table 1); DIRE syndrome was defined as immune-related events post-immunotherapy, newly incident beyond two elimination half- lives (t 1/2) of drug.
Results: We identified 10 cases, 6 by literature review (5 melanoma, 1 cutaneous SCC) and an additional 4 cases at our institution (4 HNSCC). Median cumulative immunotherapy exposure was 4 doses (range: 2 to 22 doses). Median interval from last immunotherapy dose to DIRE onset was 5 months (range: 2 to 28 months). All literature cases were in the recurrent/metastatic context; we report four cases in the curative-neoadjuvant context (italicized) with one recurrence.
Conclusions: An influx of neoadjuvant clinical trial design over the last 2-3 years, incorporating brief IO exposure (typically checkpoint blockade) followed by surgical resection and/or adjuvant therapy, is attracting interest in multiple tumor types in the curative setting.[5–9] In this context, it will be necessary to recognize an emerging phenomenon, which we have termed DIRE syndrome (delayed immune-related events). Clinical vigilance has the potential to reduce morbidity from delayed diagnosis, as these conditions are generally manageable with prompt initiation of treatment; or from misdiagnosis, to avert unnecessary/harmful interventions (in the autoimmune meningitis case we report, an Omaya reservoir was placed at an out-of-state hospital based on erroneous diagnosis of leptomeningeal carcinomatosis). Several factors confound diagnosis in the neoadjuvant-IO context: 1) intervening treatments with potentially overlapping toxicities; 2) brief and remote IO exposure; 3) reduced vigilance during NED surveillance, in contrast to active disease follow-up; 4) protracted process of diagnosis-by-exclusion. DIRE syndrome should therefore figure prominently in the differential diagnosis of patients presenting with diseases of unclear etiology, irrespective of elapsed post-immunotherapy interval.
References:
- Champiat S, et al. Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016;27:559–574.
- Abstracts of the 8th World Congress of Melanoma, the 9th Congress of the European Association of Dermatology (EADO), the 7th Interdisciplinary Melanoma/Skin Cancer Meeting, and the 3rd European Post-Chicago Melanoma Meeting. July 17-20, 2013. Hamburg, Germany. J. Dtsch. Dermatol. Ges. J. Ger. Soc. Dermatol. 2013;JDDG 11 Suppl 7:1–119.
- Parakh S, Cebon J, Klein O. Delayed autoimmune toxicity occurring several months after cessation of anti-PD-1 therapy. The Oncologist. 2018;23:849–851.
- Wang L, et al. Timing of onset of adverse cutaneous reactions associated with programmed cell death protein 1 inhibitor therapy. JAMA Dermatol. 2018; doi:10.1001/jamadermatol.2018.1912
- Forde P, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med. 2018;378:1976–1986.
- Uppaluri R, et al. Neoadjuvant pembrolizumab in surgically resectable, locally advanced HPV negative head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2017;35:6012–6012.
- Ferris R, et al. LBA46An open-label, multicohort, phase 1/2 study in patients with virus-associated cancers (CheckMate 358): Safety and efficacy of neoadjuvant nivolumab in squamous cell carcinoma of the head and neck (SCCHN). Ann. Oncol. 2017;28.
- Necchi, A. et al. Interim results from PURE-01: A phase 2, open-label study of neoadjuvant pembrolizumab (pembro) before radical cystectomy for muscle-invasive urothelial bladder carcinoma (MIUC). J. Clin. Oncol. 2018;36:TPS533-TPS533.
- Powles, T. et al. A phase II study investigating the safety and efficacy of neoadjuvant atezolizumab in muscle invasive bladder cancer (ABACUS). J. Clin. Oncol. 2018;36: 4506–4506.
-
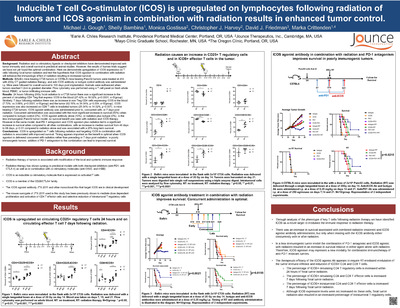
 is upregulated on lymphocytes following radiation of tumors and ICOS agonism in combination with radiation results in enhanced tumor control by Michael J. Gough, Shelly Bambina, Monica Gostissa, Christopher Harvey, David Friedman, and Marka R Crittenden")
Inducible T cell Co-stimulator (ICOS) is upregulated on lymphocytes following radiation of tumors and ICOS agonism in combination with radiation results in enhanced tumor control
Michael J. Gough, Shelly Bambina, Monica Gostissa, Christopher Harvey, David Friedman, and Marka R Crittenden
Background: Radiation and co-stimulatory ligands or checkpoint inhibitors have demonstrated improved anti-tumor immunity and overall survival in preclinical animal studies. However, the results of human trials suggest we have not yet found the optimal combination. Here we demonstrate upregulation of ICOS expression on T cells following focal tumor radiation and test the hypothesis that ICOS agonism in combination with radiation will enhance the immunologic effect of radiation resulting in increased survival.
Methods: BALB/c mice bearing CT26 tumors or C57BL/6 mice bearing Panc02 tumors were treated at d14 with 20Gy CT guided radiation therapy and anti-ICOS antibody or isotype control antibody was administrated i.p. Mice were followed for overall survival to 100 days post implantation. Animals were euthanized when tumors reached 1.2cm in greatest diameter. Flow cytometry was performed using a T cell panel on fresh whole blood, PBMC, or tumor infiltrating immune cells.
Results: 24 hours following 20Gy focal radiation to a CT26 tumor there was a significant increase in the percent of circulating CD4 Treg that express ICOS in the blood (27.42% vs 18.02%, p
Conclusions: ICOS is upregulated on T cells following radiation and targeting ICOS in combination with radiation is associated with improved survival. Timing appears important as the benefit is optimal when ICOS agonism is delivered concurrent with radiation rather than preceding or 7 days post-radiation. In poorly immunogenic tumors, addition of PD-1 antagonism to the combination can lead to improved survival.
Ethics Approval: Animal protocols were approved by the Earle A. Chiles Research Institute IACUC (Animal Welfare Assurance No. A3913-01). All experiments were performed in accordance with relevant guidelines and regulations.
-

Single cell sequencing to identify TCRs that recognize autologous tumor cells after vaccination with allogeinic DRibble vaccine
Hong-Ming Hu, Christopher C. Paustian, Zhifa Wen, Tarsem L. Moudgil, Traci L. Hilton, Sam Bookhardt, Guangjie Yu, Eric Tran, Venkatesh Rajamanickam, Walter Urba, Rachel E. Sanborn, and Bernard A. Fox
Background: Adoptive immunotherapy with tumor-specific TCR gene-modified T cells has the potential to eradicate bulky disease. Traditional methods of TCR identification require lengthy in vitro culture to generate clonal T-cell populations, which adds time and complexity to this promising therapy. Here we described a simplified and reliable method to identify TCRs by single cell TCR sequencing of cells sorted with antibodies against T-cell surface markers that are up-regulated only when they are stimulated with specific tumor cell antigens.
Methods: A tumor-infiltrating lymphocyte (TIL) culture with T cells reactive against autologous tumor was generated from a brain metastasis of a patients with NSCLC. A panel of antibodies against T-cell surface antigens was screened to identify markers that are specifically up-regulated after stimulation with autologous tumors but not with related allogeneic tumor cells. Tumor-specific T cells were sorted from TIL with three suitable antibodies and expanded by a rapid expansion protocol. Expanded T cells were examined for their tumor-specificity and subjected to single cell TCR sequencing using the 10X genomic system. The top 10 TCRs were identified by bio-informatics approach and the corresponding alpha and beta chains were synthesized and cloned into a retroviral vector based on MSG backbone. PBMC from healthy donors were transduced with the retrovirus supernatant after activation. Tumor- reactivity of transduced T cells was determined after expansion in media supplemented with IL-2, IL-7, and IL-15. To identify tumor-specific TCRs in PBMC from the same patient after vaccination with allogeneic DRibbles, we also developed a protocol to expand tumor-specific T cells from PBMC with in vitro stimulation with DRibble- loaded PBMC.
Results: We identity CD94, CD137(4-1BB), CD355 (CRTAM) as specific markers for antigen-specific activation of T-cells by autologous tumor cells, whereas other “check point” markers such as CTLA-4, PD-1, Tim3, CD39, CD103 were up-regulated by stimulation with unrelated tumor cells. These antibodies were successfully used to sort and enrich tumor-specific T cells. The top 10 TCRs from each sorting were different but with overlapping clones. Five TCR clones were tumor-specific and capable to recognize the autologous tumor cells when they were expressed on T- cells from health donors. Additionally, ex-vivo culture of vaccine stimulated PBMC from a post-vaccine timepoint generated T cells enriched for activity against autologous tumor.
Conclusions: We developed a simplified work flow to identify tumor-specific TCRs. This flow will be further improved with antibody with DNA bar codes and used to identify tumor-reactive TCRs in a streamlined fashion.
Acknowledgements: The study was supported by Providence Portland Medical Foundation and NCI SBIR grant R44 CA121612.
Ethics Approval: The study was approved by EACRI Institutution‘s Ethics Board, approval number IRB: PDX06-108
-

Pegzilarginase in combination with agonist anti-OX40 therapy enhances T cell priming and effector function leading to improved tumor regression and survival
Melissa Kasiewicz, Annah Rolig, Elizabeth Sturgill, Mark Badeaux, Scott Rowlinson, and William L. Redmond
Background: Tumor cells defective in enzymes required for arginine biosynthesis are dependent upon arginine uptake from the environment. Extracellular depletion of arginine directly affects tumor cells, inducing autophagy and apoptosis. Pegzilarginase (AEB1102) is a bioengineered, pegylated, human arginase 1 (Aeglea Biotherapeutics) currently in phase I clinical trials. This arginine-depleting agent has been shown to both inhibit arginine auxotrophic tumor growth and to enhance the efficacy of PD-L1 blockade in preclinical models. In the current study, we investigated the therapeutic efficacy and mechanism of action of combined pegzilarginase/anti-OX40 (aOX40) immunotherapy. We hypothesized that pegzilarginase/aOX40 treatment would synergize to enhance T cell priming and effector function leading to improved tumor regression and survival.
Methods: Efficacy studies were conducted in CT26 (colon) or MCA-205 (sarcoma) tumor-bearing mice. Eight days after subcutaneous tumor implantation, mice were treated with pegzilarginase (3 mg/kg; q7dx4; ip) and/or aOX40 mAb (10 mg/kg; d8, d12; ip). Seven days post-treatment (d15), the phenotype and effector status of T cells and myeloid populations within the lymph nodes (LN) and tumor were evaluated by flow cytometry. In additional cohorts, gene expression profiling (single cell RNAseq; scRNAseq) was performed 3 days post-treatment (d11). Survival studies were conducted in both models with tumor measurements taken twice weekly until tumor burden was greater than 150mm2. Data represents the results of 2-3 independent experiments (n=10/group) and for phenotyping assays, significance was determined by using a one-way ANOVA with a p-value cut-off of 0.05.
Results: We observed a significant reduction in tumor growth and increased overall survival following pegzilarginase/aOX40 therapy versus monotherapy in CT26 (p
Conclusions: Collectively, these data demonstrate that pegzilarginase in combination with OX40 agonists can significantly impair tumor growth while promoting both T cell proliferation and effector function. These insights support further exploration of this novel combination approach in future clinical trials.
-

Open-source digital image analysis of whole-slide multiplex immunohistochemistry
Nikhil Lonberg, Carmen Ballesteros-Merino, Shawn Jensen, and Bernard A Fox
Background: Successful digital image analysis (DIA) of cancer tissue is accurate and reproducible. These points of emphasis have brought procedures like the tissue microarray (TMA) and hotspot regions of interest (ROI) under scrutiny. The nature in which a pathologist selects TMAs and ROIs is conducive to bias. Whole Slide Imaging (WSI) offers a solution in its unbiased region selection and consideration of a larger tissue sample. However, options for softwares that can handle such large throughput are scarce. Additionally, while multiplex immunohistochemistry (mIHC) is becoming popular [1], documentation of its digital analysis tools remains minimal [2]. The combination of these procedures potentiates a deeper understanding of the tumor microenvironment. This study presents the whole-slide mIHC analysis capabilities of QuPath, an open-source application developed at Queen’s University Belfast [3].
Methods: A multiplex fluorescent stain panel was performed on patient samples. The slides were imaged and cells were detected and segmented in QuPath. QuPath parallelizes its workload to manage whole-slide throughput efficiently. Custom scripts were written that exhibit machine-learning and thresholding techniques to aggregate cell phenotype totals. Additionally, cell detection numbers were generated for specific ROIs and compared to a commercial DIA software. All scripts and protocols in this study are made public for replication and improvement by the community.
Results: QuPath’s automated cell segmentation and classification were demonstrated as a proof-of-concept for whole-slide multiplex immunohistochemistry analysis. Across an entire slide, cells positive for multiple markers were effectively segmented and properly phenotyped.
Conclusions: Open-source applications have become a driving force for innovation and collaboration in the field of digital image analysis. In litigating the strengths and weaknesses of QuPath for whole-slide mIHC analysis, we aim to advance the field’s knowledge of available software tools and bring attention to necessary points of growth in this rapidly changing industry.
References: 1. Feng Z, Jensen SM, Messenheimer DJ, Farhad M, Neuberger M, Bifulco CB, Fox BA. Multispectral imaging of T and B cells in murine spleen and tumor. J Immunol. 2016;196:3943-3950.
2. Blom S, Paavolainen L, Bychkov D, Turkki R, Mäki-Teeri P, Hemmes A, Välimäki K, Lundin J, Kallioniemi O, Pellinen T. Systems pathology by multiplexed immunohistochemistry and whole-slide digital image analysis. Sci Rep. 2017; 7:1-13.
3. Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, James JA, Salto-Tellez M, Hamilton PW. Qupath: open source software for digital pathology image analysis. Sci Rep. 2017; 7:1-7.
-

Preliminary evaluation of a novel whole slide multispectral assessment of seven markers: Potential to minimize bias in the characterization of the tumor immune environment
Carmen Ballesteros Merino, Shawn Jensen, Carla Coltharp, Kristin Roman, Chichung Wang, Nikhil Lonberg, Sebastian Marwitz, Tarsem Moudgil, William Miller, William Redmond, Yoshinobu Koguchi, Carlo Bifulco, Clifford Hoyt, and Bernard A. Fox
Background: PD-L1 expression and tumor-mutational burden enrich for patients that respond to checkpoint blockade, but these evaluations are only a component of the entire story. Recently, our lab reported that evaluation of specific cell-cell relationships provided a powerful biomarker for overall survival in patients with HPV- head and neck cancer (HNSCC). However, the areas selected for analysis were operator selected “hot spots”. This approach introduces the potential for unconscious bias in the selection process. To address this, we have sought to perform whole slide evaluations of sections to compare with hot spot analysis. This study is a preliminary report applying a novel set of fluorophores and filters that allow the visualization of seven colors on a whole slide.
Methods: Tissue samples included pellets of cultured lymphocytes and tumor specimens. A sample of the cultured lymphocytes that were fixed and embedded were analyzed by flow cytometry for immune markers. Formalin-fixed paraffin embedded (FFPE) sections were stained with antibodies for CD8, CD68, FoxP3, PD-1, PD-L1, cytokeratin and DAPI. PerkinElmer Opal reagents were used to identify markers and included standard and a new set of fluorophores that included Opal 480, 520, 570, 620, 690, and 780. Slides were imaged using a new scanning approach on a Vectra Polaris (PerkinElmer, Inc, Waltham, MA).
Results: Preliminary comparison of cells that were used to produce FFPE blocks by flow cytometry and multiplex IHC provided similar results for some markers. Determining optimal staining, exposure times and thresholds for analysis for this new method needs work, but the potential exists for effective evaluation of a whole slide with 7 different markers.
Conclusions: Our preliminary results provide reason to be optimistic that this approach can assess 7 colors in a whole slide. Whole sections labelled with 7 colors and spectrally unmixed supports deeper analysis of immune-biology on multiple scales, including re-analysis of spatial metrics based on emerging hypotheses about how cellular and expression distributions relate to disease progression and response to therapy.
-

Tumor infiltrating lymphocyte recruitment after peri-lymphatic IRX-2 cytokine immunotherapy in resectable breast cancer and head and neck carcinoma
Joanna Pucilowska, Venkatesh Rajamanickam, Nikki Moxon, Monil Shah, Maritza Martel, Alison Conlin, James E. Egan, and David B. Page
Background: The IRX-2 biologic is a subcutaneous injectable immunotherapy composed of IL-2 and other cytokines derived from stimulated lymphocytes. Preclinically, IRX-2 activates T cells, natural killer cells, macrophages, and dendritic cells, and facilitates maturation of antigen-presenting cells.Tumor-infiltrating lymphocytes (TILs) are associated with improved outcomes in many cancers including early stage breast cancer (ESBC) and head and neck squamous cell carcinoma (HNSCC). We report data on TIL recruitment associated with pre-operative IRX-2 in a phase Ib ESBC trial, as well as phase Ib and IIa HNSCC trials.
Methods: The pre-operative IRX-2 regimen was evaluated in both ESBC and HNSCC trials for safety and immunologic activity. Beginning 21 days prior to surgical resection, enrolled operable patients with resectable stage I-III ESBC and stage II-IVA HNSCC received single low-dose intravenous cyclophosphamide (300 mg/m2 to facilitate T-regulatory cell depletion), followed by 10 days of subcutaneous injections of IRX-2 (1mL × 2 directed to regional peri-lymphatic space, 230IU/day). Endpoints included feasibility, TIL count by H&E blinded pathology review, and Nanostring RNA analysis.
Results: In the ESBC trial, 16 patients were enrolled and evaluable for TIL analysis, whereas in the HNSCC trials, 40 patients were enrolled and 36 patients were evaluable. In both trials, all patients received all planned injections with no treatment-related surgical delays, complications, or treatment-related grade III/IV toxicities. Treatment was associated with a mean 116% relative increase in TILs (range –36% to +1275%, p = 0.02) in ESBC and a mean 58% relative increase (range -57 to +452%, p=0.01) in HNSCC. Treatment was associated with PD-L1 RNA upregulation in EBSC (mean +54%, range –53% to +185%, p=0.04) but not HNSCC, however PD-L1 was higher at baseline in HNSCC. RNA analysis in ESBC and HNSCC revealed concordant increases in cytokine gene expression, including CXCL2, CCL4, CXCR4, and CXCL12 as well as transcription factors including FOS, ETS1, NFKB, EGR1/2 which are involved in T-cell activation and differentiation. We also note augmentation of ITGAE (CD103), a known marker of memory T-cell activation in EBSC cohort.
Conclusions: Pre-operative IRX-2 was well tolerated in both tumor histologies with statistically significant TIL recruitment, as well as PD-L1 upregulation in ESBC. Future directions include an evaluation of neoadjuvant IRX-2 with anti-PD-1 and chemotherapy in stage II-III TNBC, ongoing follow-up of a randomized phase IIb trial of neoadjuvant IRX-2 regimen in HNSCC to ascertain clinical benefit, and trials evaluating efficacy of IRX-2/anti-PD-1 combination across various metastatic cancers.
Trial Registration: NCT02950259, NCT02609386
-
 and NKTR-262 (TLR7/8 agonist) combination treatment pairs local innate immune activation with systemic CD8+ T cell expansion to enhance anti-tumor immunity by Annah S. Rolig, Daniel Rose, Saul Kivimäe, Deborah Charych, Werner Rubas, Jonathan Zalevsky, and William L. Redmond")
NKTR-214 (CD122-biased agonist) and NKTR-262 (TLR7/8 agonist) combination treatment pairs local innate immune activation with systemic CD8+ T cell expansion to enhance anti-tumor immunity
Annah S. Rolig, Daniel Rose, Saul Kivimäe, Deborah Charych, Werner Rubas, Jonathan Zalevsky, and William L. Redmond
Background: Radiation therapy (RT) remains the standard of care for many human cancers. Combining NKTR-214, a CD122-biased cytokine agonist conjugated with releasable polyethylene-glycol (PEG) chains, with local RT significantly enhanced therapeutic efficacy in preclinical models. Mechanistically, NKTR-214 provides sustained signaling through the IL-2 receptor pathway (IL-2Rβγ) to preferentially activate and expand effector CD8+ T and NK cells and RT modulates the tumor microenvironment (TME) to induce antigen-release. Together, NKTR-214/RT treatment resulted in improved therapeutic responses compared to either treatment alone. However, abscopal responses in murine tumors were modest, leading us to explore alternative approaches with the potential to elicit more robust tumor-antigen specific responses. In the current study, we evaluated the extent to which NKTR-262, a polymer-modified TLR7/8 agonist prodrug, modulates the TME and synergizes with NKTR-214 treatment. We hypothesized that NKTR-214/NKTR-262 immunotherapy would promote synergistic activation of immunostimulatory innate immune responses along with systemic adaptive anti-tumor responses to significantly improve abscopal responses, tumor regression, and overall survival.
Methods: Tumor-bearing mice (CT26; 4T1) received NKTR-214 (0.8 mg/kg; iv), RT (16 Gy x 1), and/or intratumoral NKTR-262 (0.5 mg/kg). The activation status of CD4+, CD8+, and NK cells in the blood, lymph node, and/or tumor (7 days post-treatment) was evaluated by flow cytometry. Effects on innate immune subsets (macrophages, monocytes) including M1/M2 polarization were evaluated by flow cytometry and immunohistochemistry (1 day post-treatment). Data represents the result of 1-2 independent experiments (n=5-14/group). For immune markers, statistical significance was determined using a 1-way ANOVA with a p-value cut-off of 0.05.
Results: NKTR-214/RT resulted in increased absolute lymphocyte counts and expression of T cell activation markers (Ki-67, PD-1, granzyme A) in the blood and tumor. Compared to NKTR-214/RT, NKTR-214/NKTR-262 resulted in significantly improved survival (p
Conclusions: Combined NKTR-214/NKTR-262 therapy induced robust anti-tumor immunity characterized by systemic CD8+ T cell expansion, enhanced intratumoral CD8+ T cell effector function, and favorable myeloid polarization resulting in improved tumor regression and tumor-free survival.
-
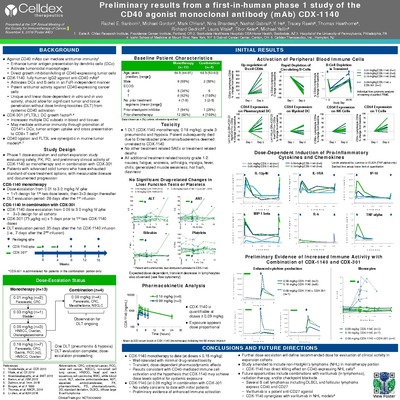
 CDX-1140 by Rachel Sanborn, Michael S. Gordon, Mark O'Hara, Nina Bhardwaj, Yi He, Tracey Rawls, Tibor Keler, and Michael Yellin")
Preliminary results from a first-in-human phase 1 study of the CD40 agonist monoclonal antibody (mAb) CDX-1140
Rachel Sanborn, Michael S. Gordon, Mark O'Hara, Nina Bhardwaj, Yi He, Tracey Rawls, Tibor Keler, and Michael Yellin
Background: Agonist CD40 mAbs can mediate antitumor immunity through multiple mechanisms, including enhancing tumor antigen presentation, activation of tumoricidal macrophages, and direct growth inhibition/killing of CD40- expressing tumor cells. To fully exploit these mechanisms may require the mAb to be dosed at levels that provide significant tumor and tissue penetration, without dose-limiting-toxicities (DLT) from systemic CD40 activation. Our agonist CD40 mAb, CDX-1140, was selected based on its unique and linear dose-dependent in vitro and in vivo activity and is postulated will achieve maximum agonist activity at dose levels associated with good systemic exposure. CDX-1140 is a fully human IgG2 agonist anti-CD40 mAb that activates dendritic cells (DCs) and B cells in an Fc receptor independent manner and has potent antitumor activity against CD40-expressing cancer cells. In addition, CDX-1140 does not block the natural CD40-CD40L interaction; combination of CDX-1140 with added soluble CD40L is synergistic in the activation of immune cells suggesting a potential to enhance in vivo CD40L dependent immune responses. In toxicology studies, CDX-1140 demonstrated potent CD40-mediated pharmacological effects without significant toxicities.
Methods: A phase 1 dose-escalation study of CDX-1140 (CDX1140-01; NCT03329950) is underway in patients with advanced tumors who have exhausted standard-of-care treatment options. The primary endpoint is determining the safety profile and maximum tolerated dose. Secondary endpoints include pharmacokinetics, immunogenicity, clinical and biological outcome assessments. Baseline and on-study biopsies will be used to explore the pharmacodynamic effects of CDX-1140 in the tumor microenvironment (TME). The dose escalation (DE) portion evaluates CDX-1140, given every 4 weeks, at doses from 0.01 to 3 mg/kg; the first 2 cohorts are single-patient cohorts and all subsequent DE cohorts are conducted utilizing a 3+3 design. Tumor-specific expansion cohorts will further explore the activity of CDX-1140. This study will also evaluate CDX-1140 in combination with CDX-301 (rhFLT3L), a DC growth factor that markedly increases DC numbers, including the CD141+ subset which are critical to an antitumor immune response and are often scarce within the TME.
Results: To date, CDX-1140 cohorts at 0.01 (n=2), 0.03 (n=1), and 0.09 (n=3) mg/kg have been completed without any drug-related serious adverse events, infusion reactions, or DLTs reported. The only drug related toxicity has been grade 1 fatigue (n=2) . Expected pharmacodynamic effects, including transient, dose-dependent decreases in lymphocyte counts and dose-dependent increases in serum IL-12p40 and TNF-Alpha, have been observed.
Conclusions: The early data suggest that CDX-1140 has the expected immune activating and safety profile.
Ethics Approval: The study was approved by University of Pennsylvania, approval number 828733; Mount Sinai School of Medicine, approval number IRB-18-00213; Providence Health and Services, approval number 201700532 and Western Institutional Review Board, approval number 115925
-

Combined anti-PD-1 and anti-LAG-3 checkpoint blockade enhances CD8+ TIL effector function while reducing Tregs leading to reduced immune suppression and improved overall survival
Elizabeth Sturgill, Courtney Mick, David Jenkins, Johanna Kaufmann, and William L. Redmond
Background: Checkpoint inhibition is a potent strategy to reinvigorate T cells. However, aCTLA-4 or aPD-1 monotherapy has not been effective for the majority of patients, resulting in the exploration of combinatorial approaches to improve treatment efficacy. One such target is LAG-3, which is upregulated on T cells that have experienced repeated antigen exposure, such as in the tumor microenvironment (TME), and is associated with reduced T cell effector function. In addition, high LAG-3 expression on regulatory T cells (Tregs) has been reported for patients with varying cancer types, providing an additional rationale for targeting LAG-3 with the aim of reducing immune suppression within the TME. We hypothesized that the combination of aPD-1 and aLAG-3 would synergize to promote tumor regression and increase survival via a reduction in tumor-induced immune suppression and enhanced CD8+ T cell effector function.
Methods: CT26 (colon carcinoma) tumor-bearing BALB/c mice received aPD-1 and/or aLAG-3 (200 μg/dose; ip) 3x/week on days 7, 10, and 13 post-tumor implant. Tumor growth (area) was assessed 2-3x/week and mice were sacrificed when tumors exceeded 150 mm2. In additional cohorts, tumors were harvested 7 days post-treatment (d17) and tumor- infiltrating lymphocytes (TIL) were analyzed by flow cytometry. Responders to combined aPD-1/aLAG3 therapy were designated as those exhibiting decreased tumor size on the day of harvest (d17) compared to maximum tumor growth post-implant.
Results: Combined aPD-1/aLAG-3 immunotherapy significantly improved the survival of CT26 tumor-bearing mice compared to monotherapy (p
Conclusions: In summary, these data suggest that aPD-1/aLAG-3 immunotherapy increased recruitment of CD8+ TIL exhibiting enhanced effector function, increased CD4+ Teff/Treg ratios, which likely mitigated Treg-mediated immune suppression. Together, these positive immunological changes led to a more immune stimulatory TME capable of supporting tumor regression and significantly improved tumor-free survival.
-

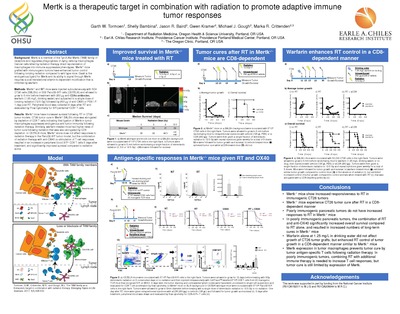
Mertk is a therapeutic target in combination with radiation to promote adaptive immune tumor responses
Garth Tormoen, Jason R Baird, Gwen Kramer, Shelly Bambina, Marka R Crittenden, and Michael J. Gough
Background: Mertk is a member of the Tyro3-Axl-Mertk (TAM) family of receptors and regulates phagocytosis of dying cells by macrophages. Cancer cells killed by radiation therapy direct repolarization of macrophages into immune suppressive phenotypes. Mertk-/- mice grafted with immunogenic tumors have enhanced tumor control following ionizing radiation compared to Mertkwt mice. Gas6 is the endogenous ligand for Mertk and its ability to signal through Mertk requires a post-translational vitamin k-dependent modification that is inhibited by warfarin.
Methods: Mertk-/- and WT mice were injected subcutaneously in the flank with 5E4 CT26 cells (BALB/c) or 5E6 Panc02-SIY cells (C57BL/6) and allowed to grow to 5 mm before treatment with 250 μg anti-CD8α antibodies, warfarin (0.5 mg/L drinking water) and subjected to a single dose of ionizing radiation (16 Gy) followed by 250 μg of OX40 or PBS I.P. 1-day post-RT. Peripheral blood was collected 6 days after RT and evaluated by Flow Cytometry for SIY- pentamer+CD8+ T cells.
Results: Radiation therapy results in tumor control in BALB/c mice, but tumor cure in Mertk-/- BALB/c mice. Tumor cure in Mertk-/- BALB/c mice was abrogated by depletion of CD8 T cells indicating that ligation of Mertk in tumor macrophages suppresses endogenous anti-tumor immunity following radiation therapy. Similarly, warfarin-treated mice had higher rates of tumor cure following radiation that was also abrogated by CD8 depletion. In C57BL/6 mice, Mertk-/- alone does not affect responses to radiation therapy in the Panc02 tumor model, but the combination of radiation therapy with anti-OX40 costimulation of T cell responses resulted in a significant increase in peripheral blood SIY+ CD8 T cells 5 days after treatment, and significantly improved survival compared to radiation alone.
Conclusions: Mertk-/- mice, and Mertkwt mice treated with warfarin to inhibit Gas6 experience increased tumor control following ionizing radiation in an adaptive-immune mediated manner in CT26 tumor models. In less immunogenic tumors, loss of Mertk-/- permitted tumor cure following radiation therapy when combined with the T cell costimulatory molecule OX40. These data demonstrate that Mertk suppresses adaptive immunity in irradiated tumors. Mertk is an attractive therapeutic target in combination with ionizing radiation and immune therapy to promote adaptive immune anti-tumor responses.
Ethics Approval: All animal studies were approved by the Earl A. Chiles Research Institute IACUC, Assurance No. A3913-01.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}